
生体内合成化学治療
〜体内で直接薬剤分子や高分子、さらに材料を化学合成してその場で治療機能を発揮させる〜
【研究の背景】がん治療の現状と抗がん剤開発の難しさ
私達の生活を脅かす様々な疾患、例えばがんに焦点を絞っても、どれくらいの方が罹患され、この病と日々闘っておられるか、申し上げるまでもありません1)。世界中の化学者が、がんを治すために有効な薬を求めて努力しています。抗がん剤を開発するときにはまず、その薬の候補化合物を作り(合成)、そして実験室で効くかどうか検討します。もし有効な薬の候補が見つかれば、さらに実験動物を使ってその効果を検証しますが、多くの場合、この段階で開発が止まってしまいます。実験室でいくら効果的であっても、その候補化合物ががん細胞に届かない、あるいは他の正常な臓器にも薬効が働き、副作用が出るなど、様々な要因で開発が止まります。1種類のがん細胞に薬を運ぶことのできる、抗体やナノ粒子がドラッグデリバリーシステムとして開発されていますが、実際の患者様のがんは「不均一」であり、多種類のがん細胞が集まってがんを形成しています。全ての患者様のがんに適応させるには、現実的ではありません。高い薬効を持つ抗がん剤を開発すると同時に、体の中で安定に、そしてがん細胞にのみ薬効を働かせなければなりません。私達は薬(分子)を作る「有機合成化学」を専門とする化学者です。近い未来になんとか「有機合成化学」の力で解決できないものか?これまでのようにフラスコで薬を開発するのではなく、「有機合成化学」の力を使った全く新しい、何らかの方法で、と常に考えてきました。私達が10年以上の間、東京科学大学(東工大)と理研で開拓してきた「生体内合成化学治療」の概略を述べるとともに、この世界で唯一の化学技術を基盤とした研究の将来展望と夢を述べさせていただきます。
体の中で抗がん剤を化学合成する「生体内合成化学治療」を臨床の現場で実現する
私達は、「生体内合成化学治療」という新概念を世界に先駆けて提出し、生きている動物の体の中で抗がん剤を作ることに挑戦してきました。これまでのように、投薬して病気を治すのではありません。抗がん剤の「もと(原料)」となる安全な分子を体内に入れ、直接がん部で薬を作って(合成)、そしてそのがん細胞にのみ薬効を効かせる技術です。体内のがん現地で直接、薬剤を調製することにより、副作用の問題を根本から解決して患者様を救う画期的なアイデアです。当初はほとんどの研究者からクレイジーなアイデアであり、決して実現はできないと酷評されましたが、10年以上に及ぶ東京科学大学(東工大)と理研の皆様のサポートと苦労の末、私達はマウスの体内で様々な「抗がん剤(分子)そのもの」を触媒的に合成して治療することに初めて成功しました2~6)。私達の革新的な化学触媒(遷移金属触媒)は、血液内でも体内の臓器内でも全く失活せず、実験室のフラスコ内と全く同じように効率的に触媒反応を起こして、様々な抗がん剤を体内で合成することが可能となりました7~12)。さらに田中研で独自に開発した「糖鎖クラスター」を体内デリバリーシステムとして使用することで、この化学触媒を血中に投与するだけで、様々なヒト患者の「不均一な」がんにも選択的に到達させることを可能としました。一旦、ほんの僅かな触媒をがんに埋め込むだけで、数日間、患者様に即したいろいろな抗がん剤をテイラーメイドに合成・量産できるようになりました13~16)。今後、薬剤耐性を持つがんが出現した場合にも、即座にそのがんに適した抗がん剤を体の中で複数種類合成し、適当な分子を体内のがんの現場で探索して治療する革新的戦略、すなわち世界初の「体内創薬ライブラリー合成・探索研究」を実現いたします。
さらに私達が次に目指しているのは、体内でポリマーを化学合成する研究です。血液に溶けないポリマーは静脈から入れると詰まってしまいますが、脳梗塞やがんの多量出血の際に、溶解性の良い低分子と触媒を注射し、疾患の現地で速やかにポリマーを合成して止血します。この挑戦的な研究によって、がんだけでなく出血で危篤な病状にある多くの患者様を助けることができると信じています。さらに近い将来には、血管の望む位置で望む機能性材料までをも触媒的に合成して、温度を測ったり、細胞を培養したり、あるいは体内で新しいエネルギーを生み出して循環させることも夢ではないと考えています。
このように、私達の世界初、そして未だ世界で唯一の体内触媒技術が起爆剤となり、田中研究室を基点として国内外で体内での有機合成化学を発展させます。さらにこれらの技術を臨床の現場に展開することにより、近未来には抗がん剤を体の中で最適化する「体内創薬研究」で医療分野の常識を変えることを目指します。
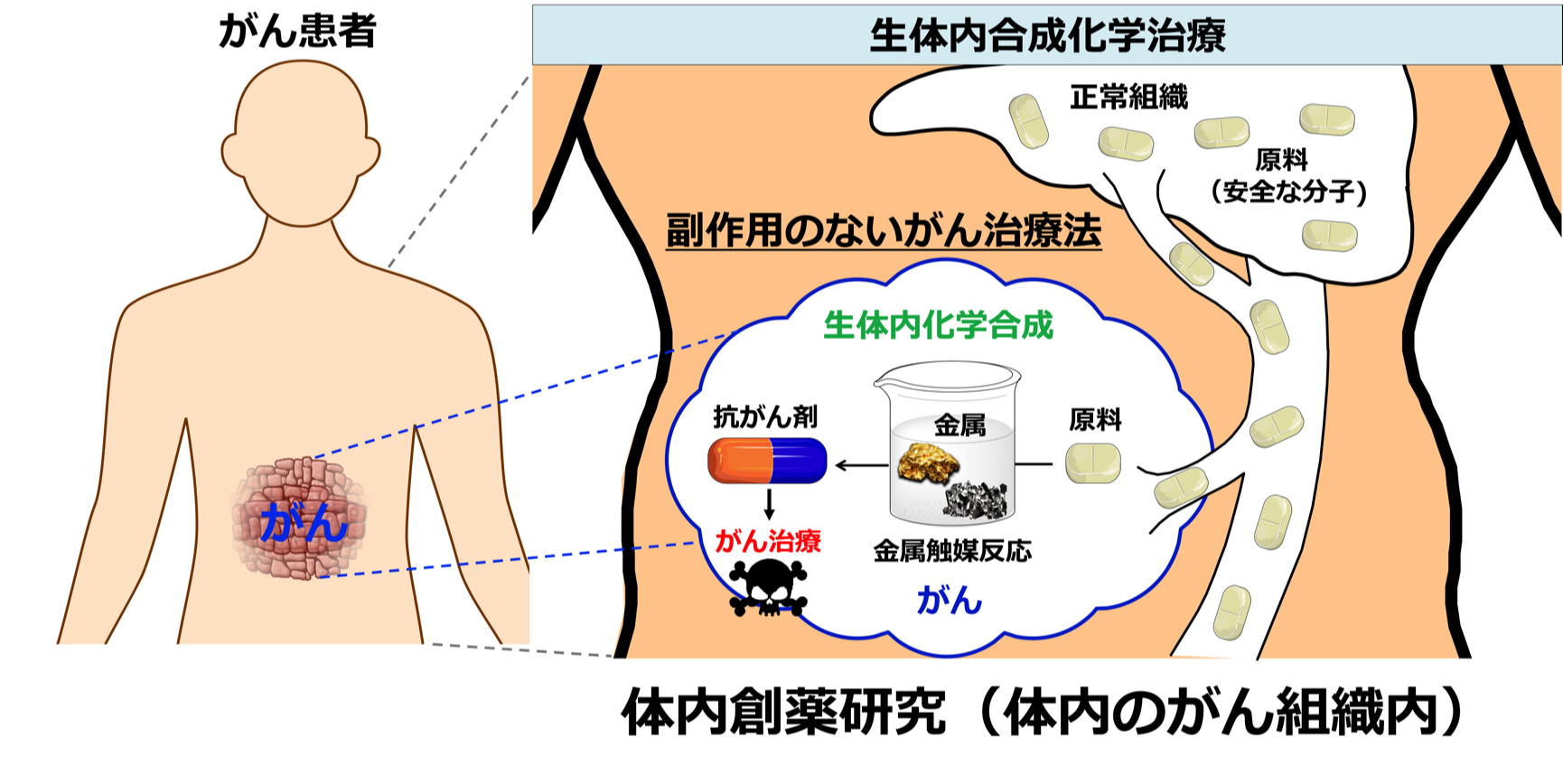
体内の疾患部位で生産される原料を駆使して効率的に「生体内合成化学治療」を実施する
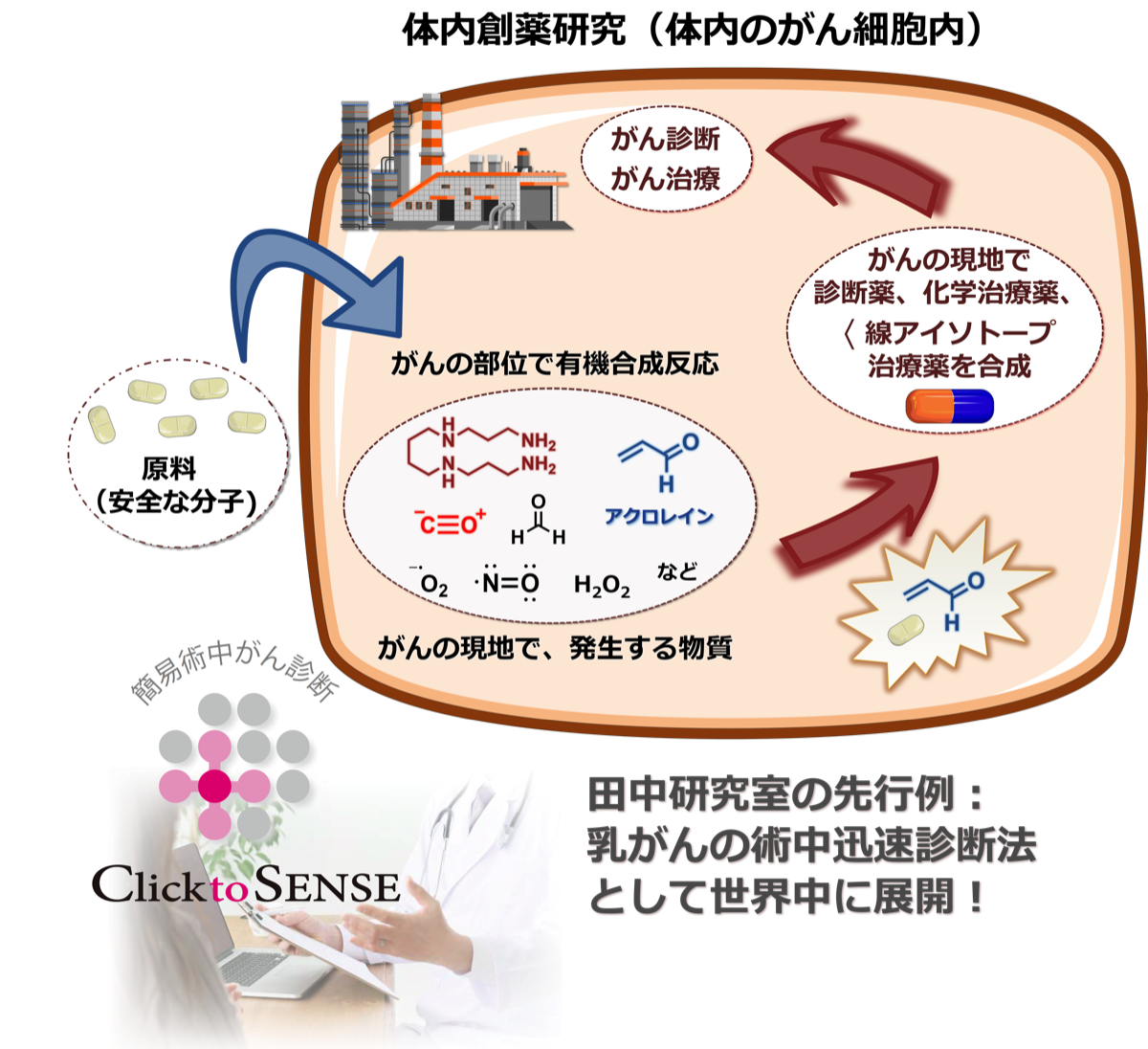
体内での創薬研究をより効率的に実施するためには、抗がん剤の原料をいつも注射しているだけではパフォーマンスが良くありません。がんの現地では、化学反応を起こせるほどの濃度で発生する化学物質が生成していますので、それを積極的に抗がん剤の原料に使用します。私達は、世界で初めて「アクロレイン」という分子ががんでのみ、時にはミリモーラーの濃度で発生していることを発見しました17)。実験室の細胞株でも、がん患者様のがんでも、がんになると必ず発生する初めてのバイオマーカーです。私達は、患者様のがんで発生するアクロレインを原料として使い、前述の体内での化学触媒反応と融合させることで、モデルマウスや実際の患者様のがんで診断薬や化学治療薬18-20)、あるいは理研の仁科加速器科学研究センターとの共同作業によりa線アイソトープ治療薬21)を現地合成して、効率的にがんを診断したり治療することに成功しています。
この一例として、大阪大学病院と共同で、乳がんの手術の時に、患者様のがんで発生するアクロレインと反応させることにより、たった数分でがんであるか、そうでないかを確実に判断する診断薬を「患者様の乳腺組織」で直接作り、どんな種類の乳がんでも97%以上の精度で見分けることに成功しました17,22)。この化学技術により、乳房を切除する際、広範囲に渡って切除するのではなく、できるだけ乳房を残したまま必要ながん部だけを取り除くことがたった数十分で可能となりました。2024年4月に関西を中心とした多施設臨床性能試験(治験)を始め、患者様にいち早くこの診断薬を届けます。さらにアクロレインを体内での原料として、患者体内での乳がん治療薬の開発と臨床展開を急ピッチで進めており、10%にもおよぶ女性の疾患を救うための一般技術として確立します。
さらに私達は、アクロレインだけではなく、がんでは別の異常代謝物が高濃度で生産されていることを見出しています。これらを体内での有機合成化学の原料として効果的に使うことにより、体内での創薬研究が活躍する時代を田中研が創っていきます。
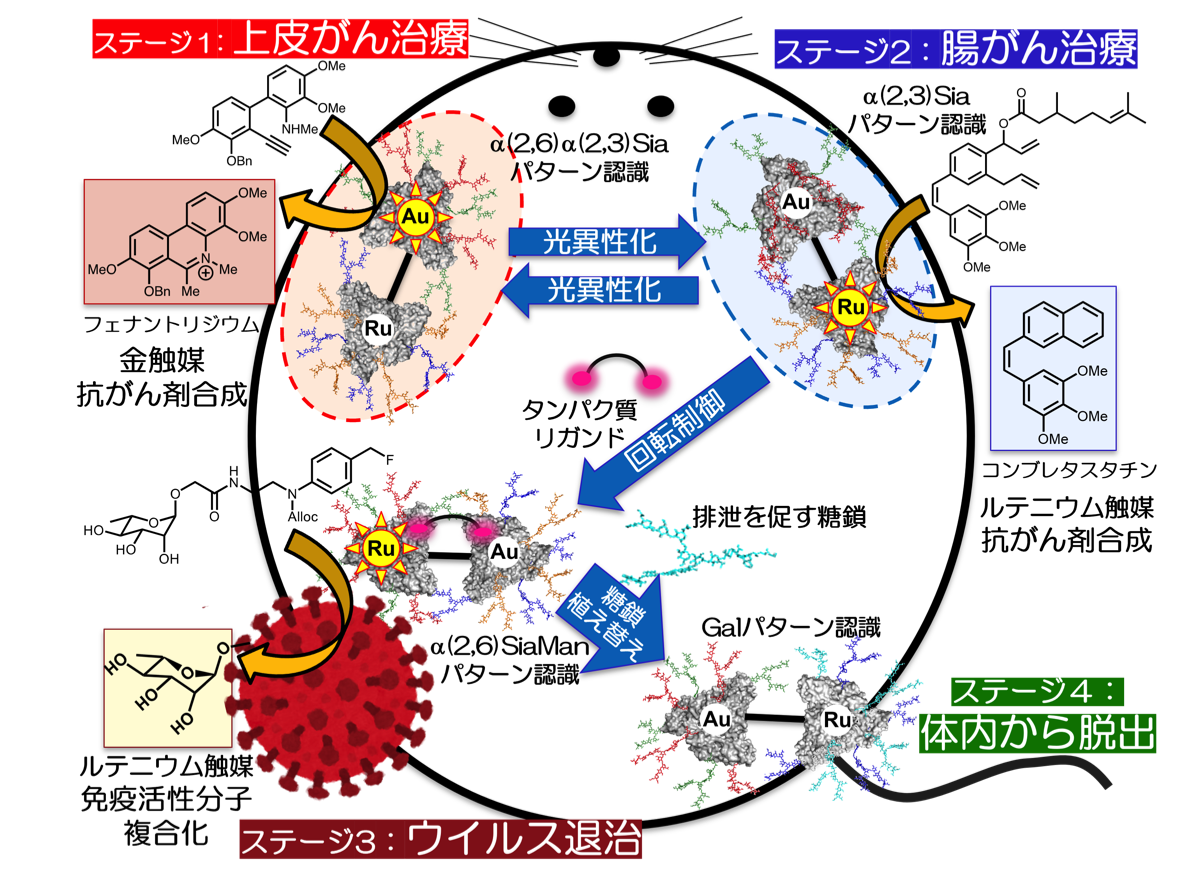
体内を循環し、疾患を見つけて治療し、最後に体内から脱出する「ミクロの決死圏」を体内での有機合成化学で実現する
さらに私達は、上記で述べた体内での創薬研究と、田中研究室で最近開発した「化学的刺激で臓器間を移動する糖鎖クラスター技術」23)を巧みに組み合わせることにより、マウス体内の疾患をパトロールして診断・治療する世界初の技術開発に挑みます。体内を循環するパトロール分子により、例えば上皮がんや大腸がん、あるいはウイルスを即座に見つけるとともに、その疾患に適した薬剤分子や機能性材料を触媒的に現地合成して効果的に治療します。治療が完了した後には、巡回させたパトロール分子を膀胱や大腸から排泄(脱出)させることにより、体内診断・治療が完結します。これまでにも、ナノ粒子に様々な薬剤を内包することで、「ミクロの決死圏」を目指した研究が行われてきましたが、このような従来型のドラッグデリバリーシステムでは一度きりの治療しかできません。体内の特定の疾患の間を自在に行き来させ、複数の疾患で複数の薬剤を触媒的に合成・生産することは不可能です。私達が独自に開発した化学的刺激や体内化学反応を駆使することにより、50年以上、ハリウッド映画の中だけでの夢であった「ミクロの決死圏」を田中研究室で初めて実現します。
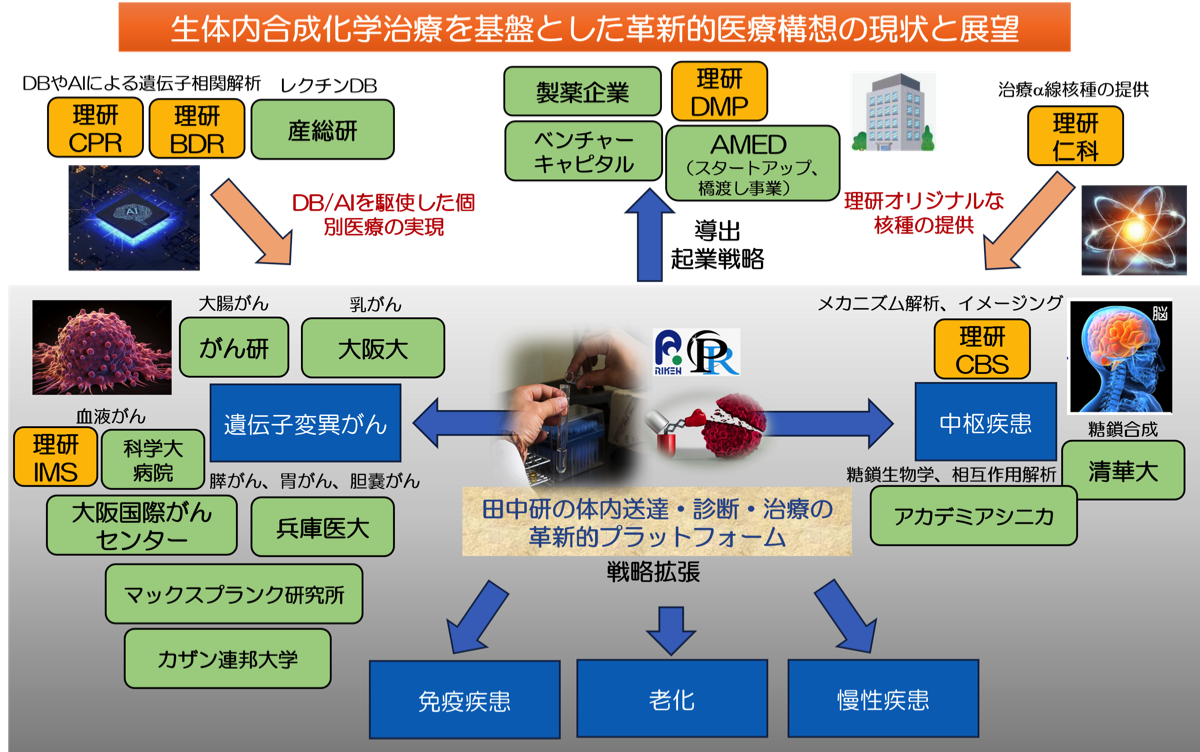
生体内合成化学治療を基盤とした革新的医療構想 〜国内外の共同研究と社会実装に向けた展望〜
田中研のアカデミア研究成果を多くの患者様に還元するために、「生体内合成化学治療」、ならびにこの戦略を成す個々の送達・診断・治療技術について、東京科学大学(東工大)や理研内外との堅牢な協力体制を構築し、田中研究室オリジナルなプラットフォームとして確立します。今後、特に理研のDMP(創薬・医療技術基盤プログラム)やベンチャーキャピタルの支援を得て、国内外の医療機関や製薬会社に展開し効果的に社会実装を実現します。
既にがん研、大阪大学病院、医科歯科大学病院、理研IMS、大阪国際がんセンター、マックスプランク研究所、ロシアカザン連邦大学(戦争により2021年に一旦停止)と協力し、特定の遺伝子変異がんやがん関連細胞、または免疫や老化細胞をターゲットとした研究を開始いたしました。さらに2024年度から、理研の戦略的パートナー連携事業プロジェクトとして、台湾の清華大学、アカデミアシニカ、さらに理研CBSと共同で神経疾患をターゲットとした研究も開始しています。これら様々な疾患細胞の遺伝子情報とレクチンデータベース(DB)を基に、機械学習から様々な疾患標的認識分子を取得する次世代技術を開拓します。取得した認識分子に体内創薬を実現する化学触媒に加え、さらに理研仁科センターのa 線や東京科学大学や理研の内外で開発されている低分子薬剤、あるいは生物製剤まで様々なペイロードを導入することにより、現在、治療手段がない患者様に対して革新的な個別治療手段を提供いたします。まさに、ライフサイエンスを変革する国際戦略的医療構想を近未来に実現したいと考えています。
【最後に】未来の化学治療 〜有機合成化学の分野から20年後に活躍する新しい技術開発を〜
私達は今後、もはや従来法を用いるだけでは良薬となりうる革新的な化合物の発見は難しいと考えています。一方で、私達の「生体内合成化学治療」により、活性が弱かったり、副作用に悩まされて治療する時代は終わり、近い将来、副作用なく的確に疾患細胞のみをターゲットとした新しい薬剤を患者様のお手元にお届けします1)。「生体内合成化学治療」は、新しい分子を開発する技術ではありません。素晴らしい活性を持つ一方で、副作用や動態が悪かったために従来、見捨てられてきた化合物の可能性を、もう一度体の中で取り戻す「有機合成化学」によるルネッサンスです。生物の分野では、mRNAを体内に導入してワクチンを開発したり、体内で免疫細胞をがんを殺傷する細胞へと変更するなど、体内での創薬研究は既に始動しています。化学分野においても、体内での創薬研究が未来の治療戦略を変えることができると信じています。困っておられる患者様を目の当たりにし、私達化学者は実験室で論文を書くだけでなく、患者様を救う体内合成化学をどんどん開拓すべきであり、これが田中克典研究室の使命であると考えています。
【参考資料】
1) 世界がん撲滅サミット: https://cancer-zero.com.(田中が2年連続で基調講演に選出)
2) Angew. Chem. Int. Ed., 56, 3579 (2017).
3) Sci. Adv., 7, eabg4038 (2021).
4) Chem. Sci. 12, 12266 (2021).
5) Nature Commun., 13, 39 (2022).
6) Chem. Sci., 14, 11033 (2023).
7) Nature Catal., 2, 780 (2019).
8) Nature Commun., 10, 5746 (2019).
9) Chem. Sci., 11, 10928 (2020).
10) Angew. Chem. Int. Ed., 60, 2 (2021).
11) Angew. Chem. Int. Ed., 61, e202205541 (2022).
12) Angew. Chem. Int. Ed., 63, e202411225 (2024).
13) Nature Commun., 14, 5803 (2023).
14) Nature Chem. Biol., 19, 1291 (2023).(エディターによる田中研の紹介記事)
15) Nature Commun., 15, 3543 (2024).
16) Nature Chem. Biol., 21, in press (2025).
17) Adv. Sci., 6, 1801479 (2019).
18) Chem. Sci., 12, 5438 (2021).
19) Chem Sci 15, 9566 (2024).
20) Chem Sci 15, 9566-9573 (2024).
21) Chem Sci 14, 8054 (2023).
22) Eur. J. Surg. Oncol., 48, 1520 (2022).
23) Nature Commun., 15, 7409 (2024).
